Protein-protein interactions are crucial for many cell processes like metabolism, signal pathways, gene control, and cell communication. While large-scale proteomics studies keep discovering more interacting proteins, only a few potential complexes can be directly studied in experiments. Understanding a protein-protein interaction's structure is key to knowing how it works and what the proteins involved actually do. But getting crystal structures of these complexes is tougher than for individual proteins, and many interactions are short-lived, making traditional study methods like crystallography and NMR challenging. That's why there's a growing need for quick and effective computational approaches to reliably predict these interaction structures.
Protein-protein docking is all about using computers to predict how proteins would form complexes as they do in a living cell, using already solved structures of the individual proteins. At Profacgen, we use top-notch docking software to figure out how two proteins might fit together to form a stable complex. The docking process has two key parts: 1) Creating a bunch of possible configurations that hopefully include one that's almost correct, and 2) Using scoring to pick out the best match. We start with a rigid body global search using Fast Fourier Transform (FFT) to explore how one protein can rotate and shift around a fixed partner. If we know where the binding sites are, we narrow down the search area. The conformations we generate are then scored and ranked based on how well they fit together, their electrostatic interactions, and how they handle desolvation. We cluster the results to find the most common low-energy shapes, and we can further refine these structures with energy minimization.
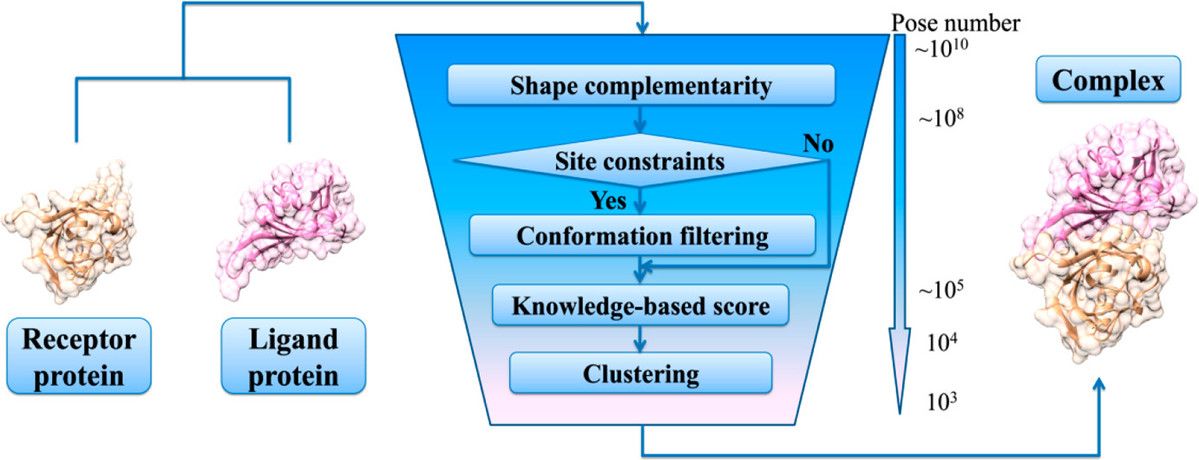 Fig1. Schematic diagram of protein docking process. (Kong R, et al., J Chem Inf Model. 2019)
Fig1. Schematic diagram of protein docking process. (Kong R, et al., J Chem Inf Model. 2019)
Understanding Disease Mechanisms
Protein-protein docking helps unravel the complex interactions behind diseases, shedding light on how proteins link to various pathologies. It provides crucial insights into potential therapeutic targets by mapping out the molecular basis of diseases. For instance, in cancer, docking studies have clarified how certain proteins interact in tumor growth. Similar approaches in neurodegenerative and infectious diseases have revealed new angles on disease progression and treatment avenues.
Drug Discovery and Development
Docking plays a significant role in designing biologic-based therapies. By helping identify novel drug targets and fine-tune drug candidates, it streamlines the process of finding effective treatments. Docking can predict how potential drugs bind to protein complexes, making the discovery process faster and less costly. This approach not only speeds up drug development but also enhances the likelihood of success by targeting the right interactions.
Basic Research and Structural Biology
In the realm of basic research, protein–protein docking is invaluable for studying the interactions within protein complexes that drive essential cellular functions. It also aids in predicting the quaternary structures of homo-oligomers, offering insights into protein assembly. Our services even support modified amino acids and membrane protein docking, highlighting the adaptability and breadth of applications in various research fields.
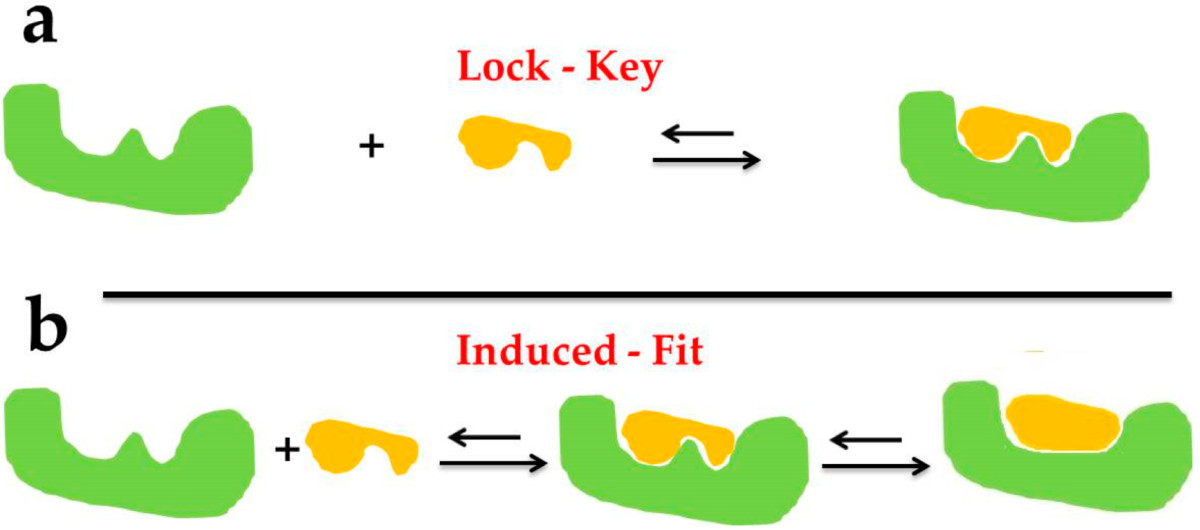 Fig2. The docking types of Lock–Key Model (a) and Induced Fit Theory (b). (Chen G, et al., Mar Drugs. 2020)
Fig2. The docking types of Lock–Key Model (a) and Induced Fit Theory (b). (Chen G, et al., Mar Drugs. 2020)
In studying protein interactions within cells, experimental approaches often hit their limits, pushing us to use computational methods like protein-protein docking to dive into these complex interactions. Profacgen offers cutting-edge protein docking services, using the latest tech and algorithms to help researchers understand how proteins interact, speeding up drug discovery and basic research. Our service is thorough and tailored, from initial consultation to delivering results, each step is designed to meet your specific needs and ensure project goals are met smoothly.
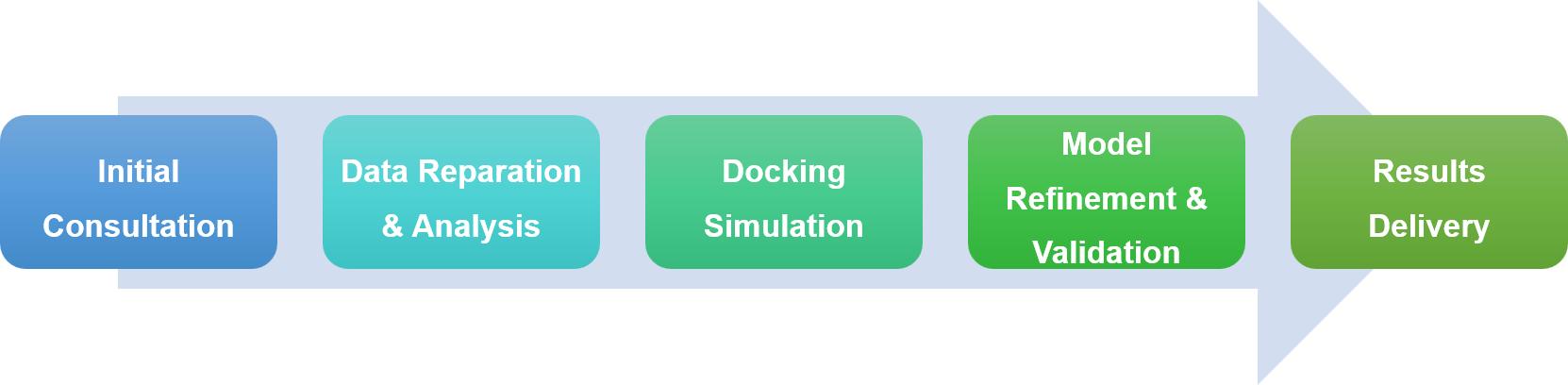
Profacgen employs some of the most advanced docking software and algorithms in the business, heavily relying on Fast Fourier Transform (FFT) correlation methods to efficiently explore the vast range of possible protein conformations. Our process begins by generating a broad set of configurations to ensure no potential fits are missed. We then score these configurations to zero in on the most stable complexes, making sure we focus on those that offer the best potential for further study.
Our services are fully customizable to meet the unique needs of each client. Whether you know specific binding sites, have desired interaction partners, or certain research questions, we adjust our methods accordingly. For those requiring more detailed insights, we offer high-resolution flexible docking, refining the structures and pinpointing hot-spot residues crucial for interactions, providing a deeper understanding of your protein complexes.
Project
Protein-Protein Docking Between Two Proteins
Background
The service quotation is for exploring the binding mode and identifying key binding site residues between two proteins, A protein and B protein. The goal is to deliver the binding mode and key binding site residues to the customer.
Methods and Materials
Based on the sequence information, the 3D structures of A protein and B protein were downloaded from the PDB database. H-DOCK was used to perform the docking calculations, and the top 10 optimal conformation models were output.
Results
The docking score of A protein and B protein was -234.34, with a confidence score of 0.8464. Protein-protein/complex docking scores are typically less than -200. When the confidence score is higher than 0.7, the two molecules are most likely to bind. When the confidence score is between 0.5 and 0.7, two molecules will likely bind. When the confidence score is below 0.5, the two molecules are less likely to bind.
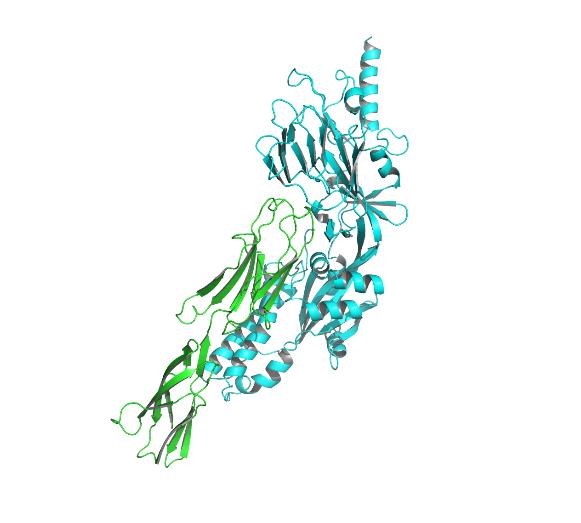 Fig3. Protein-protein docking based on a hybrid algorithm of template-based modeling and ab initio free docking.
Fig3. Protein-protein docking based on a hybrid algorithm of template-based modeling and ab initio free docking.
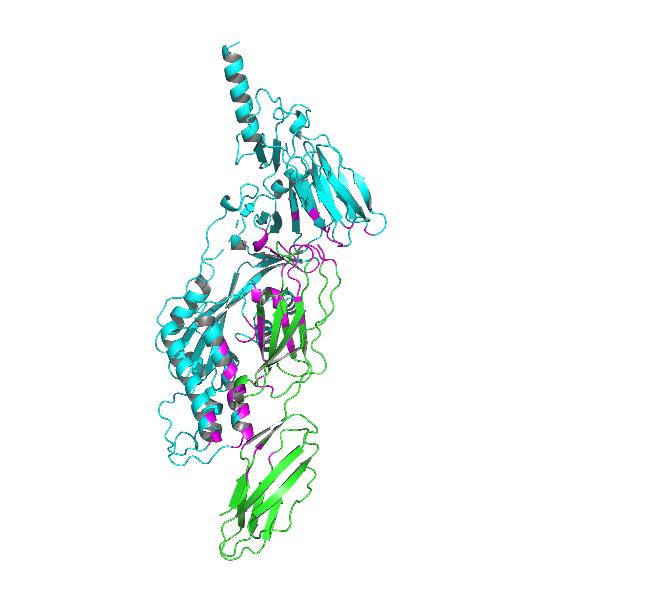 Fig4. Docking model of ICM-1 protein and A&B protein.
Fig4. Docking model of ICM-1 protein and A&B protein.
A protein interacts with B protein mainly through hydrogen bonds, hydrophobic interactions, and salt bridges. The specific interactions are as follows:
Table 1. Details of Hydrogen Bonds
| A Amino Acid | B Amino Acid |
| GLU160 | THR249 |
| GLN89 | GLU218 |
| THR172 | GLN283 |
Multiple amino acids of the A protein have hydrophobic interactions with the amino acids of B protein.
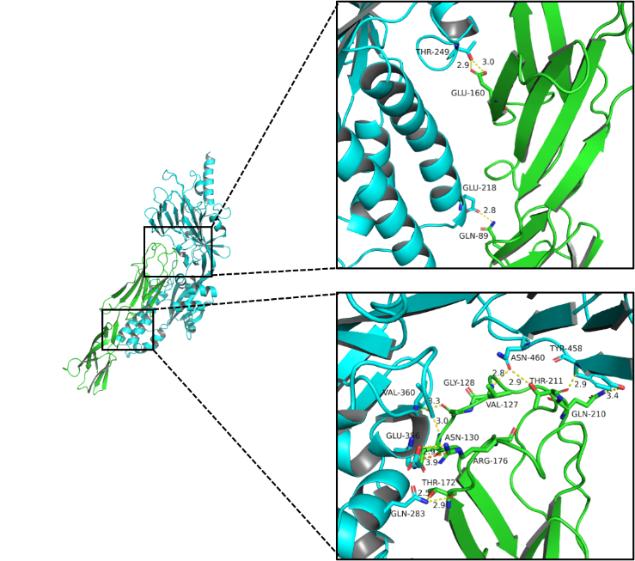 Fig5. 3D detail map.
Fig5. 3D detail map.
Conclusions and Discussions
The project successfully completed the interaction analysis between A and B proteins. The calculated results indicated that it is highly likely that there is an interaction between these two proteins. The detailed interaction analysis provided insights into the binding mode and key residues involved in the interaction, which can be useful for further experimental validation and drug design.
| Protein Structure Modeling | Molecular Dynamics Simulation | Bioinformatics Data Management | Protein Interaction Analysis Services | A New Platform for Protein High-Throughput Screening |
References:
Fill out this form and one of our experts will respond to you within one business day.