The interaction between proteins and their cognate ligands plays an important role in many essential biological processes and metabolic pathways, including signal transduction, transport, cell regulation, gene expression control, and enzyme inhibition. Many experimental techniques are now available for the detection and measurement of these binding interactions, but it remains difficult and expensive to obtain complex structures by experimental methods, such as X-ray crystallography or NMR. Thus, computational docking is considered an important approach for study of protein-ligand interactions and for drug discovery and development.
Profacgen makes use of the most state-of-the-art protein–ligand docking software tools to predict the position and orientation of a ligand when it is bound to a protein receptor by calculating the site, geometry and energy. The process of docking a ligand to a binding site mimics the natural course of interaction of the ligand and its receptor via the lowest energy pathway. Typically our modeling procedures starts with a target of known structure, such as a crystallographic structure of a protein of interest. Docking is then used to predict the bound conformation and binding free energy of small molecules to the target. Every docking protocol can be described as a combination of a search algorithm and a scoring function. The search algorithm generates a large number of poses of a small molecule in the binding site, allowing the degrees of freedom of the protein–ligand system to be sampled sufficiently as to include the true binding modes. The scoring function calculates the score or binding affinity of a particular pose, which represents the thermodynamics of interaction of the protein–ligand system, in order to distinguish the true binding modes from all the others explored, and to rank them accordingly.
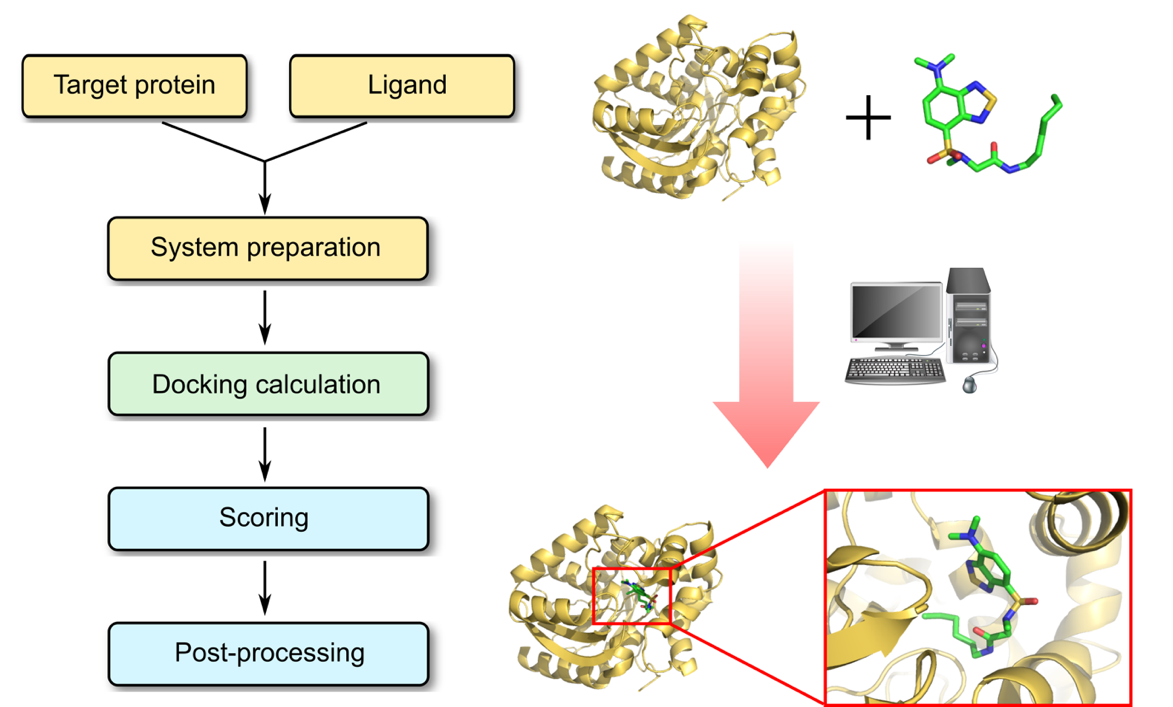
Profacgen employs docking techniques for a variety of purposes. Single docking experiments are useful for exploring the function of a protein, studying enzyme inhibitors and substrates, elucidating biochemical pathways. Most notably, docking can be applied to the virtual screening of large databases of available chemicals for lead detection and optimization, which offers unparalleled opportunities for structure-based drug design and discovery.
We provide the service in a customizable fashion to suit our customers’ specific research goals. Please do not hesitate to contact us for more details about our protein–ligand docking service.
Fill out this form and one of our experts will respond to you within one business day.