Protein–Lipid Docking
Membrane proteins play critical roles in a wide range of cellular functions. The attachment of proteins to biological membranes has shown to regulate cell signaling and many other important cellular events through a variety of mechanisms. In vivo interactions with membrane lipids help stabilize the structure of these proteins, and promote rearrangement, assembly, dissociation, or conformational changes within many protein structural domains, resulting in an activation of their biological activity. Some cytosolic proteins are recruited to different cellular membranes via specific membrane-targeting structural domains. Besides, lipids are also the natural substrates of many important enzymes in key metabolic pathways. Therefore, it is of critical importance to understand the interactions between proteins and lipids.
Protein–lipid interactions are stabilized by the formation of intermolecular hydrogen bonds, van der Waals interactions, hydrophobic interactions, and ionic bridges (especially aspartate or glutamate residues) between the protein and lipid ligand. Profacgen makes use of the state-of-the-art computational software tools to predict these protein–lipid interactions. The lipid binding sites of a protein can be deduced from its amino acid sequence, and/or predicted from its three-dimensional structure using molecular docking protocols. Our docking method combines sequence and structure information, and explores the most energetically favorable protein-lipid complex. The scoring function is specifically designed to allow for the prediction of lipid distortion and protein conformational changes associated with the binding event. Experiment-derived restraints can also be applied to limit the search space. The structures with best values of binding energies are clustered and representatives of the largest clusters are selected for structural optimization by energy minimization before being presented to the customer. The stability of docked complexes can be further tested through molecular dynamic simulations.
Protein–Lipid docking process
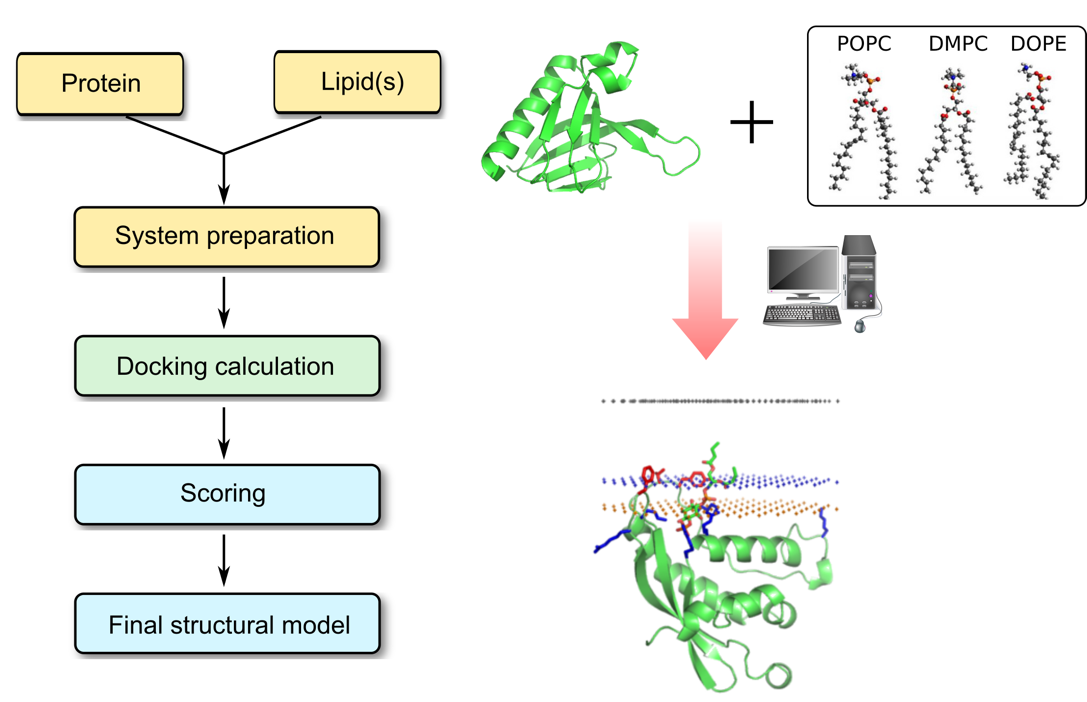
Interactions with lipids have been shown to be important for the structure and function of a variety of membrane proteins. Our computational prediction is an important means for identifying lipid binding region in a protein and understanding the physicochemical forces that underlie specific protein–lipid interactions at the atomic level. The final predicted structure also provides a model of protein/lipid interactions which can form a starting point for more detailed atomistic MD simulations and/or detailed biophysical investigations.
Features
Statistical potentials dedicated to assessing protein–lipid interactionsSupport for various membrane binding mechanisms: covalent, hydrophobic, electrostatics, hydrogen bonds, van der Waals interactions
Selection of appropriate lipid type and composition (with support for up to 32 different lipid types)
Simulation of effect on membrane distortion and protein structural change
Customizable to support single detergent or micelles
Seamless integration with downstream MD simulations
We provide the service in a customizable fashion to suit our customers’ specific research goals. Please do not hesitate to contact us for more details about our protein–lipid docking service.