Chromatin Immunoprecipitation (ChIP) Assay Service
Background
Chromatin immunoprecipitation (ChIP) is a molecular biology technique used to investigate protein-DNA interactions and identify binding sites within the genome for specific proteins. Through ChIP, researchers can understand how transcription factors bind to gene promoter regions, how histone modifications affect chromatin accessibility and gene expression, and how non-coding Rnas interact with chromatin. With the development of technology, derivative technologies have emerged, providing higher throughput and more accurate analysis methods.
Chromatin immunoprecipitation-sequencing (ChIP-seq) is a high-throughput sequencing technique used to study protein-DNA interactions. It combines ChIP techniques with Next-Generation Sequencing (NGS) techniques. In the physiological state, the DNA in the cell is cross-linked with the protein after cracking the cell, separating the chromosomes, and randomly cutting the chromatin by ultrasound or enzyme treatment. The DNA fragments bound to the target protein are then precipitated using antigen-antibody specific recognition reactions. Then the DNA fragments of binding proteins were released by backcrosslinking, and the target DNA fragments were purified and the library was constructed. Finally, the information of protein-DNA interaction was obtained by high-throughput sequencing.
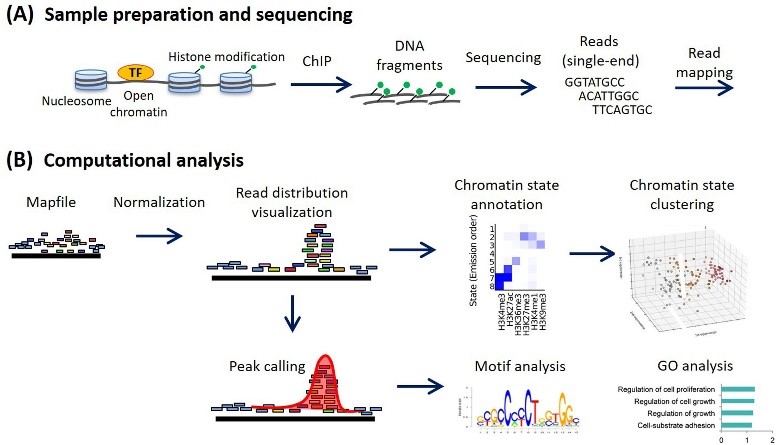 ChIP-seq analysis workflow. (Ryuichiro Nakato, 2021)
ChIP-seq analysis workflow. (Ryuichiro Nakato, 2021)
ChIP technology is widely used in molecular biology and epigenetics research, allowing scientists to explore the interactions between proteins and DNA.
 Transcription factor analysis.
Transcription factor analysis.
By using antibodies that target specific transcription factors, ChIP can reveal the precise binding sites of these transcription factors in the genome, and thus understand how they regulate gene expression. Involved in orchestrating epigenetic disorders that cause cancer, autoimmune diseases, allergies, and many other diseases.
 Epigenetic modification analysis.
Epigenetic modification analysis.
ChIP technology can be used to analyze various post-translational modifications of histones, such as acetylation, methylation, etc., which are closely related to chromatin status and gene expression regulation.
 Analysis of chromatin structure and status.
Analysis of chromatin structure and status.
Acetylation or methylation on specific histone residues can affect gene activation or silencing as well as higher-order chromatin structure. Through ChIP, these histone modification features can be used to predict those properties of epigenetic regulation in specific genomic regions.
 Study of disease-related mechanisms.
Study of disease-related mechanisms.
ChIP technology is also very important in the study of disease mechanisms, which can reveal changes in gene regulatory networks under disease states. It can also be used to study how drugs work by influencing how specific proteins interact with DNA.
Service Procedure

Platforms
Bio-RAD CFX 96 Real-Time PCR Systems
Use the Bio-RAD CFX 96 fluorescence quantitative PCR instrument to accurately quantify the effective concentration of the library (the effective concentration of the library is > 10 nM) to ensure the quality of the library.

Illumina NextSeq 500 Platform
Qualified libraries were sequenced on the Illumina NextSeq 500 platform and 150 bp paired-end reads were generated.

Analysis pipeline
After obtaining the sequencing data (Raw Data), it is first necessary to filter the data to obtain high-quality data (Clean Data). And compare the clean data with the reference genome to get the BAM file. Then conduct a genome-wide peak scan, and analyze the differential genes by GO analysis, KEGG pathway analysis, motif analysis, and visualization.
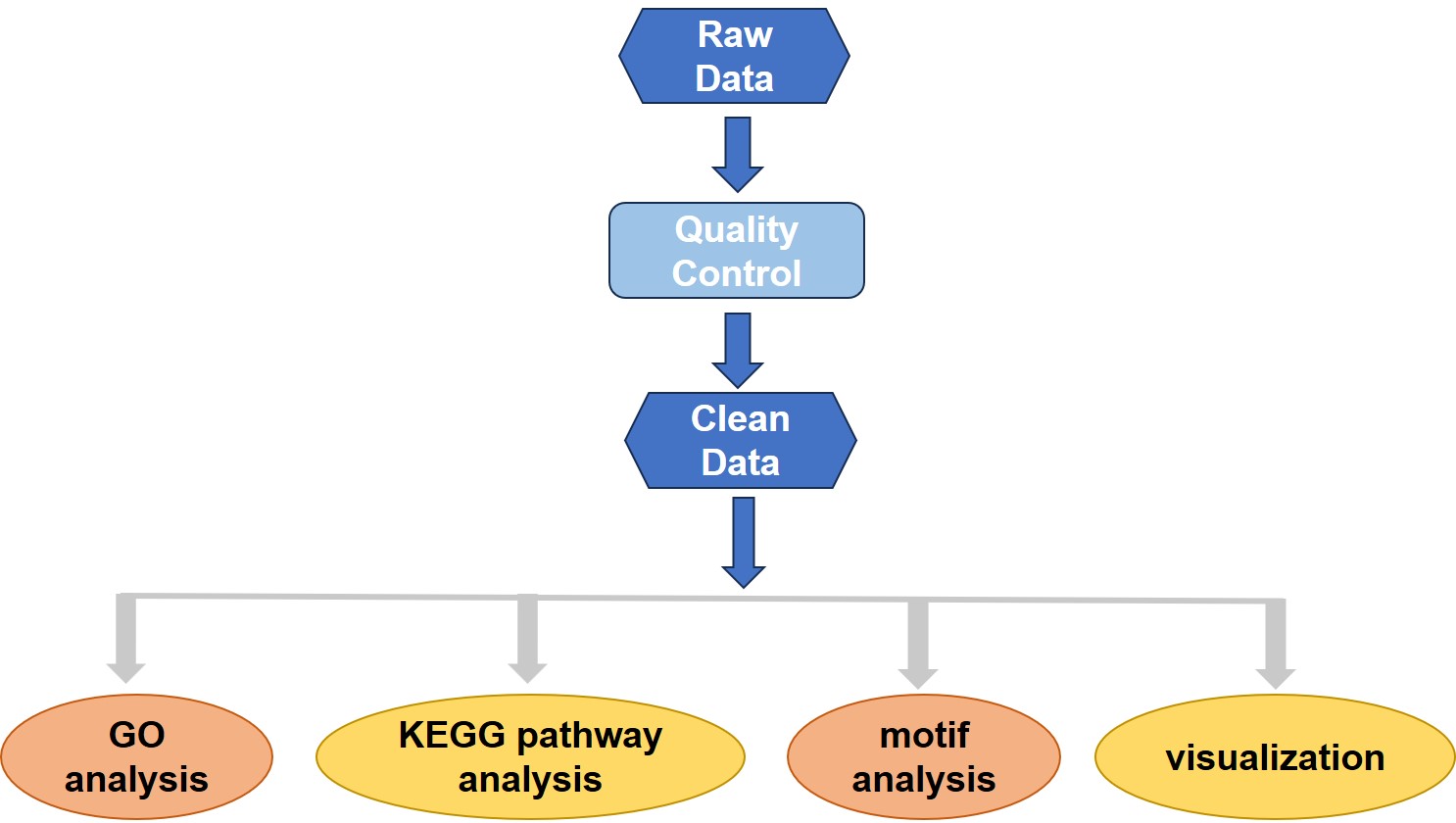
Our Advantages
- One-stop service
- Professional bioinformatics analysis
- High resolution and high signal-to-noise ratio results
- Best price in market
Case Study
Background
ChIP Assays is based on the cells to be analyzed. After received cells from the customers, Profacgen started with cell culture and fixation, followed by chromatin digestion, protein-DNA complex immunoprecipitation, Library constriction, sequencing, and bioinformatics analysis. Through all the above steps, we completed the ChIP-seq analysis. With the clean data, we can provide comprehensive data analysis including GO analysis, KEGG pathway analysis, motif analysis, and visualization. Below show some results in the ChIP Assay project.
Results
1. Enrichment-related gene functional enrichment (GO) analysis
Gene Ontology can be divided into three parts: molecular function (Molecular Function), biological process (Biological Process) and cellular component (Cellular Component). ChIP assay provides direct evidence of gene regulation and can identify binding sites on the genome where specific transcription factors or histone modifications are made. GO analysis allows functional annotation of genes near these binding sites, revealing the role of these genes in biological processes, molecular function, or cell composition through enrichment analysis. GO analysis will return a p-value for the GO of each gene, and a small p-value indicates that the differential gene is enriched in the GO.
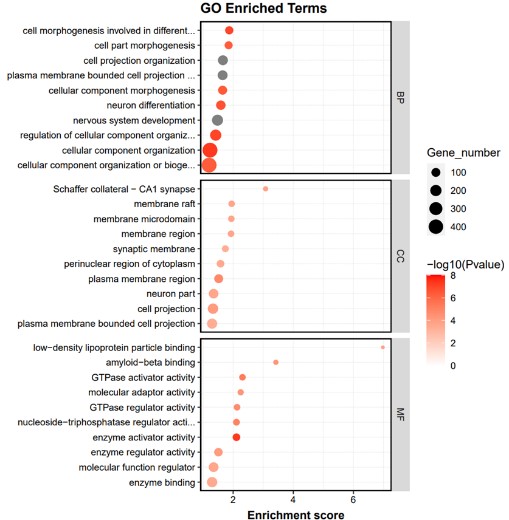 Fig1. Result of GO enrichment analysis.
Fig1. Result of GO enrichment analysis.
2. Biological pathway (KEGG) analysis
After ChIP assay provides the binding sites of transcription factors or histone modifications on the genome, KEGG analysis uses this information to further investigate how genes near these binding sites participate in known biological metabolic pathways and signaling pathways through pathway databases, contributing to a more comprehensive understanding of the molecular mechanisms of gene expression regulation. KEGG analysis was performed on the found Peak-related genes, from the perspective of complex regulatory networks, based on the KEGG biological pathway database (http://www.genome.jp/ ), the KEGG database-based biological pathway enrichment analysis was performed on the differential peak-related gene sets.
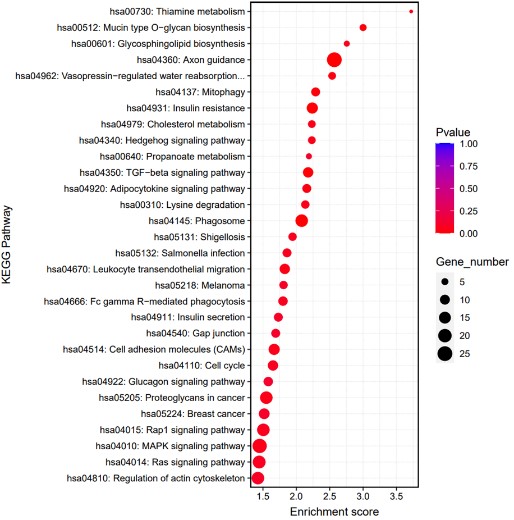 Fig2. Result of KEGG pathway analysis.
Fig2. Result of KEGG pathway analysis.
3. Motif analysis
Motif analysis can be used to verify potential binding sites found in ChIP assay, confirm the biological significance of experimental results by searching the moscriptions of known transcription factors, and achieve functional prediction of potential regulatory factors. Use Homer software to perform motif analysis on the DNA in the region where the peak is located. Customers can view the motif enrichment analysis results through the homerResults.html file.
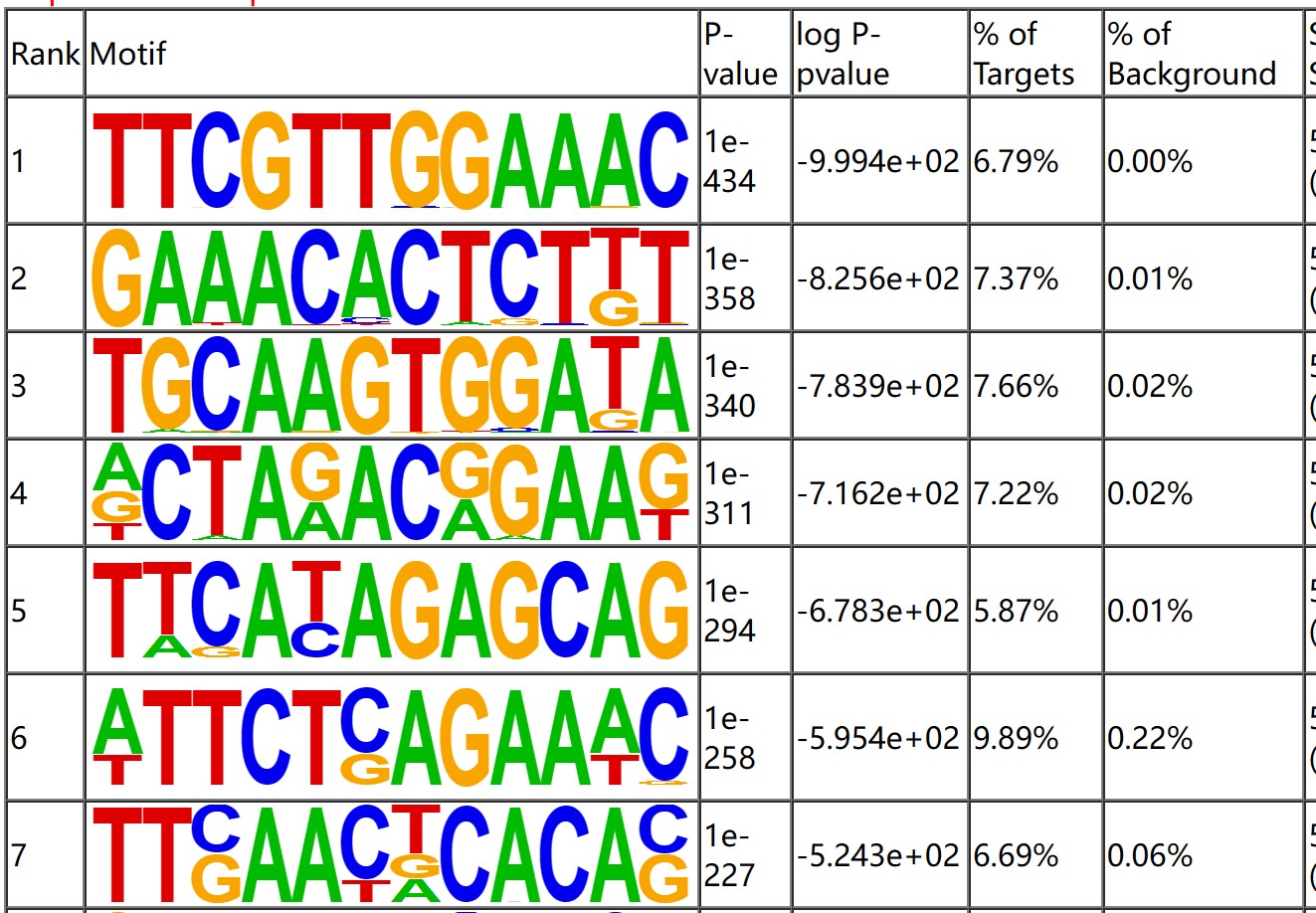 Fig3. Result of Motif analysis.
Fig3. Result of Motif analysis.
FAQs
References:
- Nakato R, Sakata T. Methods for ChIP-seq analysis: A practical workflow and advanced applications. Methods. 2021;187:44-53.
- Ashburner M, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25-29.
- Ogata H, et al. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27(1):29-34.