The interactions between protein and nucleic acids play essential roles in various essential biological processes, including DNA replication, RNA transcription, RNA splicing, degradation of nucleic acids and protein synthesis. Defects in protein–nucleic acid interactions are implicated in a number of diseases, ranging from neurological disorders to cancer. Our understanding of these processes will improve as new structures of protein–nucleic acid complexes are solved and the structural details of the interactions are analyzed. However, experimental determination of most protein–nucleic acid complex structures by high-resolution methods is a tedious and difficult process.
Computational techniques complement experimental approaches in elucidating protein–nucleic acid interactions. In silico docking of proteins with nucleic acids, by building theoretical models of the complex structures at atomic details, can yield sufficient information to build a working hypothesis and guide further experimental analyses to identify important amino acids or nucleotide residues.
Profacgen makes use of state-of-the-art docking software tools to find the relative transformation and conformation of the protein and nucleic acid involved in energetically favorable complex formation. The general procedures are: First, a rigid body global search is performed and geometrically plausible protein–nucleic acid complex structures are generated. In this process, user-defined restraints can be applied to limit the search space. The resulting models are scored and ranked using statistical potentials, developed specifically for protein–RNA or protein–DNA complexes. The best-scored structures are then clustered and representatives of the largest clusters are selected for structural optimization by energy minimization before being presented to the customer.
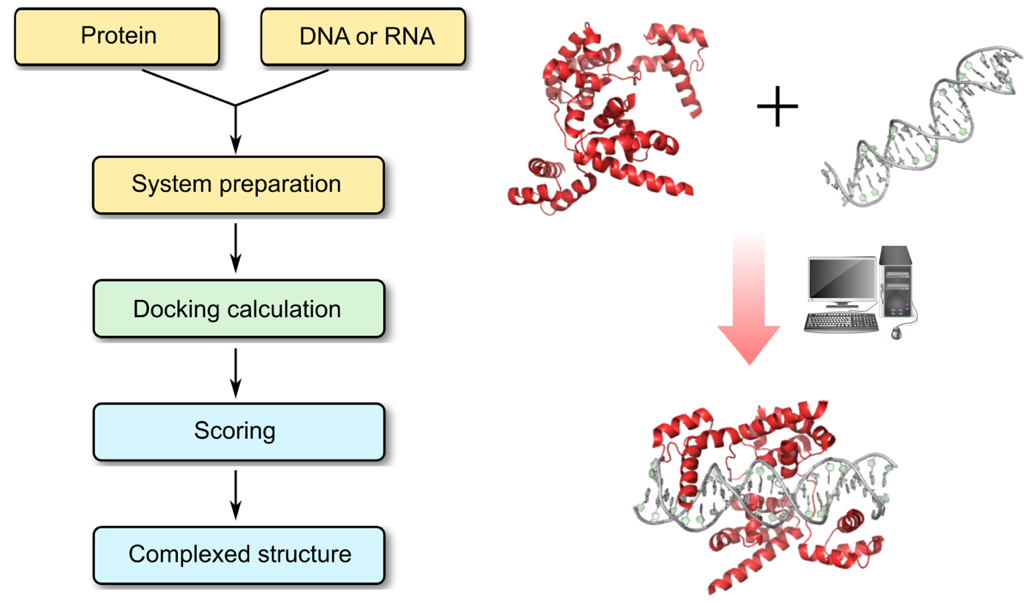
Profacgen employs computational docking techniques to search all possible binding modes in the translational and rotational space between the protein and nucleic acids (DNA, RNA, or hybrid DNA/RNA), which can undergo very large conformational changes upon complex formation. This is an important means for understanding the physicochemical forces that underlie macromolecular interactions and a valuable tool for modeling protein–nucleic acid complex structures at the atomic level. Furthermore, the precise understanding of these interactions involving disease-implicated targets is ever more critical for the rational design of biologic-based therapies.
We provide the service in a customizable fashion to suit our customers’ specific research goals. Please do not hesitate to contact us for more details about our protein–nucleic acid docking service.
Fill out this form and one of our experts will respond to you within one business day.