Knowledge about the tertiary structure of a protein is critical for understanding of its biological function and for the discovery of novel therapeutics. Computational prediction of protein structure is a powerful alternative and supplementary approach to experimental methods such as X-ray crystallography, NMR and cryo-EM. An important task in protein structure prediction is to identify proteins that have similar tertiary structures, which can be used as a template to model the unknown structure of another protein. The process for identifying these structurally similar proteins and is called fold recognition (or threading), a useful method for predicting the structure of a query protein, especially when the query protein shares a low sequence level identity (i.e. <25%) with other proteins with known structure.
Profacgen provides our expertise in helping recognize the correct structural fold among known template protein structures for a target protein by combining sequence profile-profile alignment with multiple structural information. Our approach starts with the construction of a structure template database/library, and then incrementally replacing the sequence of a known protein structure in a library with a query sequence of unknown structure. Alignment of the target sequence with each of the structure templates is achieved by optimizing the designed scoring function. The process is repeated against all known 3D structures in the template database until an optimal fit is found. The most statistically probable alignment is selected as the threading prediction and a structural model is constructed for the target by placing the backbone atoms of the target sequence at their aligned backbone positions of the selected structural template. Unlike sequence-only comparison, these methods take advantage of the extra information made available by 3D structure information. The resultant structural models are all quality validated and can be used downstream in silico or experimental work such as protein engineering and drug design.
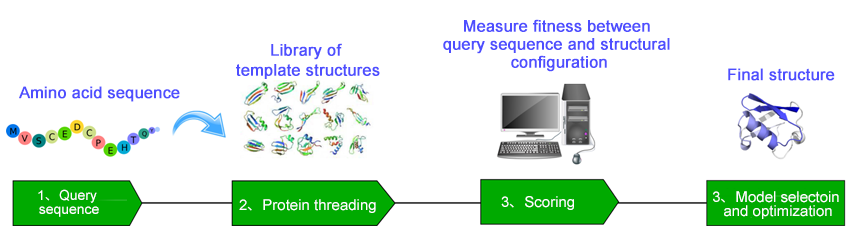
The output includes information about template proteins, the query-template alignments and most importantly full-length models of the target protein. We can also customize the service according to the specific requirements from our customers and integrate our computational procedures into your workflow. Please do not hesitate to contact us for more details about our fold recognition service.
Fill out this form and one of our experts will respond to you within one business day.