The process of drug discovery involves the identification of molecular candidates, synthesis and test of chemical compounds for their pharmaceutical efficacy. Computer-Aided Drug Design (CADD) is a specialized sub-discipline of rational drug design that uses computational methods to investigate/predict drug-receptor interactions. Quantum mechanics (QM) is an essential tool in CADD research. High-throughput in silico screening of ligand binding (such as docking or QSAR) can significant reduce the time required for compound discovery and optimization. However, these rapid methods often lack the accuracy in exploring the binding mechanism details. On the contrary, we can obtain a more accurate representation of molecular systems with QM approaches.
Profacgen provides QM calculation service to describe the protein system, including ligands and solvent, in order to gain a better understanding of protein-ligand interactions with improved accuracy. The utility of QM can provide accurate force field parameters from results of ab initio calculations on small model structures. It can also explicitly describe the electronic structure of a molecule.
Modeling studies on drug interactions with biological targets such as receptors, enzymes or other bio-macromolecules involve large chemical systems. In molecular mechanics, molecules are described with ball-and-spring models, and the forces can be calculated from potential energy functions that considers structural properties such as bond lengths, etc.
Calculating the properties for a large chemical library using QM would be time consuming and cost prohibitive. High accuracy QM/MM calculations are multi-scale computational methods to study ligand binding. Using the combination of quantum chemistry that represents the ligand and molecular mechanics that characterizes the protein and solvent, the modeling time can be drastically reduced without sacrificing the accuracy of results in most cases.
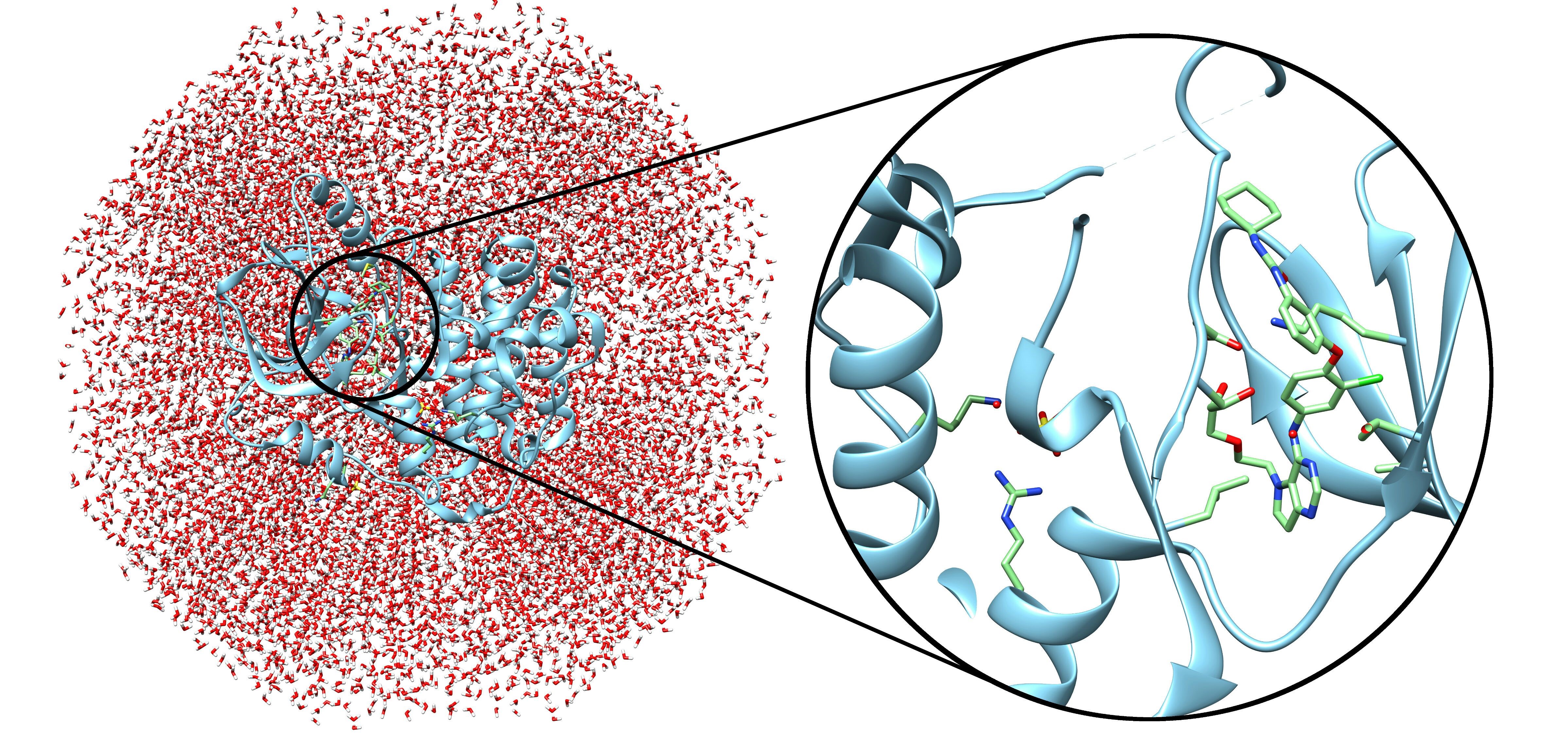
Profacgen enables the efficient use of QM with modern quantum molecular algorithms that allow efficient searching of the chemical spaces. Our optimized protocol utilizes high performance computing techniques to provide faster and more accurate prediction.
The service of QM calculations is highly customizable according to the specific requirements from the customers. Please do not hesitate to contact us for more details about our computer-aided drug design service.
Fill out this form and one of our experts will respond to you within one business day.